Alzheimer and Parkinson diseases are the most common and famous neurodegenerative disorders. However, most neurodegenerative disorders are rare and largely disregarded by the pharmaceutical industry. We are developing molecular tools and cellular models to study some of them, such as Huntington and Alexander diseases, ARSACS and SPAX8.
Huntington´s disease
Huntington’s disease (HD) is a neurodegenerative disorder characterized by the loss of medium spiny neurons in the striatum. The main histopathological hallmark of HD is the misfolding and subsequent intracellular aggregation of a mutant form of huntingtin (HTT). HTT is a very large protein (~350 kDa), but expression of exon 1 is sufficient to produce HD-like features in various cellular and animal models. HTT exon 1 (HTTex1) contains a polyglutamine (polyQ) tract that, in normal conditions, is constituted by 6 to 35 glutamine residues (Fig. 1). An expansion of the polyQ tract beyond 35 glutamines induces the misfolding and aggregation of mutant HTT and causes HD. Mutant HTT with longer polyQ expansions is more prone to aggregate, and leads to earlier onset of the disease.
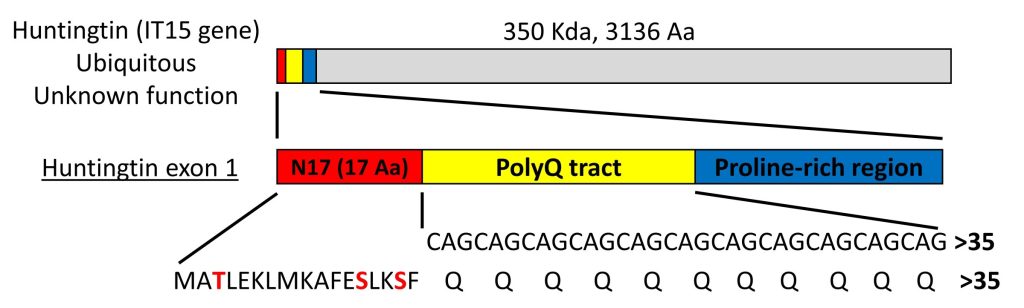
The polyQ tract is preceded by an N-terminal sequence of 17 amino acids (N17 domain) that is highly conserved, suggesting that this domain plays an important role in the function of HTT (Fig. 1). The N17 domain plays a key role in the aggregation pathway of HTT, where the protein associates first into alpha-helical oligomers and acts as a seed to concentrate and facilitate the formation of larger aggregates and fibrils. Deletion or posttranslational modifications such as phosphorylation, ubiquitination and SUMOylation in the N17 produce striking effects in the stability and aggregation of HTT, as well as in cell viability. The N17 domain has 3 phosphorylatable amino acid residues – threonine at position 3 (T3), and serine residues at positions 13 and 16 (S13 and S16)(Fig. 1). Phosphorylation of any of these residues prevents HTT aggregation in vitro and in vivo, and most often exerts neuroprotection in animal models of HD. We are interested in how these and other N17 post-translational modifications affect the behaviour of HTT in living cells.
For more details:
Branco-Santos J, Herrera F*, Pires-Afonso Y, Poças GM, Giorgini F, Domingos PM*, and Outeiro TF* (2017). Protein phosphatase 1 regulates huntingtin exon 1 aggregation and toxicity. Hum Mol Genet. 2017 Oct 1;26(19):3763-3775. doi: 10.1093/hmg/ddx260. *, Co-corresponding author.
Alexander disease
Autosomal Recessive Spastic Ataxia de Charlevoix-Saguenay (ARSACS)
Spastic Ataxia 8 (SPAX8)